
As plaquetas, também conhecidas como trombócitos, são fragmentos celulares encontrados no sangue. Elas desempenham um papel crucial no processo de coagulação sanguínea, que é essencial para interromper sangramentos após lesões vasculares. As plaquetas são pequenas, não possuem núcleo e têm uma vida útil curta, geralmente em torno de 7 a 10 dias.

Função das Plaquetas:
Quando ocorre uma lesão em um vaso sanguíneo, as plaquetas são ativadas e aderem à área danificada. Elas liberam substâncias químicas que atraem mais plaquetas para o local, formando um agregado plaquetário. Esse agregado, também conhecido como tampão plaquetário, ajuda a selar a lesão e inicia o processo de coagulação.
Produção e Liberação:

A produção e liberação de plaquetas são estimuladas principalmente pelo hormônio trombopoietina, que é produzido no fígado e rins em resposta às necessidades do organismo. Quando há demanda aumentada por plaquetas, a trombopoietina estimula a produção dessas células na medula óssea, assegurando uma resposta eficaz para situações de lesões vasculares ou hemorragias. Esse processo é parte de um sistema complexo de regulação, envolvendo outros fatores e citocinas, que mantêm o equilíbrio necessário para a homeostase no sistema circulatório.
- Estímulo Inicial:
- A necessidade de mais plaquetas no organismo pode ser desencadeada por situações como lesões vasculares, hemorragias ou outras demandas hemostáticas.
- Produção de Trombopoietina:
- O fígado e os rins produzem e liberam a trombopoietina em resposta a essa demanda aumentada.
- Ação da Trombopoietina:
- A trombopoietina atua nos megacariócitos, células precursoras encontradas na medula óssea. Ela estimula a produção e a maturação dessas células.
- Formação de Plaquetas:
- Os megacariócitos sofrem um processo de fragmentação, liberando plaquetas na corrente sanguínea.
- Circulação de Plaquetas:
- As plaquetas circulam pelo sangue, prontas para responder a qualquer lesão vascular.
- Feedback Negativo:
- À medida que a contagem de plaquetas aumenta, há um feedback negativo que regula a produção de trombopoietina para evitar uma produção excessiva.
- Outros Fatores de Regulação:
- Além da trombopoietina, outros fatores e citocinas, como interleucinas, também desempenham papéis na regulação da produção de plaquetas.
- Manutenção da Homeostase:
- O equilíbrio delicado entre a produção e a remoção de plaquetas assegura que o sistema circulatório mantenha a homeostase, respondendo eficientemente às necessidades hemostáticas do corpo.
Cascata de coagulação:
A cascata de coagulação é um processo complexo que envolve uma série de reações bioquímicas e ativação de fatores de coagulação para formar um coágulo sanguíneo. Essa cascata é essencial para prevenir sangramentos excessivos quando ocorrem lesões nos vasos sanguíneos. A cascata de coagulação é frequentemente dividida em três fases: a via intrínseca, a via extrínseca e a fase comum.
Via Intrínseca:
- Ativação Inicial:
- Lesões nos vasos sanguíneos expõem o colágeno, ativando a via intrínseca. O fator XII (Hageman) é ativado em contato com a superfície lesada.
- Cadeia de Ativação:
- O fator XII ativa o fator XI, que, por sua vez, ativa o fator IX em conjunto com o fator VIII ativado pela trombina. A ativação ocorre em uma série de reações em cadeia.
- Ativação da Tromboplastina Parcial Ativada (TTPa):
- O TTPa é prolongado nesta fase, indicando a eficácia da via intrínseca.
Via Extrínseca:
- Estímulo Externo:
- Lesões externas que extravasam o sangue ativam a via extrínseca. O tecido danificado libera o fator tecidual (tromboplastina).
- Ativação do Fator VII:
- A tromboplastina forma um complexo com o fator VII, ativando-o.
- Ativação da Tromboplastina Total (TP):
- O TP mede a eficácia da via extrínseca e é frequentemente avaliado clinicamente.
Fase Comum:
- Ativação do Fator X:
- Nas convergências das vias intrínseca e extrínseca, o fator X é ativado.
- Formação da Protrombinaase:
- O fator X ativado, em conjunto com o fator V ativado pela trombina, converte a protrombina em trombina.
- Conversão de Fibrinogênio:
- A trombina converte o fibrinogênio em fibrina.
- Estabilização do Coágulo:
- A fibrina forma uma rede que estabiliza o coágulo sanguíneo.
Fibrinólise:
- Dissolução do Coágulo:
- A fibrina é gradualmente dissolvida pela plasmina, iniciando o processo de fibrinólise.
A cascata de coagulação é finamente regulada para evitar coágulos inadequados ou sangramentos excessivos. Desregulações nesse sistema podem levar a distúrbios hemorrágicos ou trombóticos. O equilíbrio delicado da cascata de coagulação é essencial para a homeostase sanguínea.
Contagem Normal de Plaquetas:
A contagem normal de plaquetas em um microlitro de sangue varia de 150.000 a 450.000. Valores abaixo desse intervalo podem indicar trombocitopenia (baixa contagem de plaquetas), aumentando o risco de sangramento. Valores acima podem indicar trombocitose (alta contagem de plaquetas), aumentando o risco de coágulos sanguíneos.
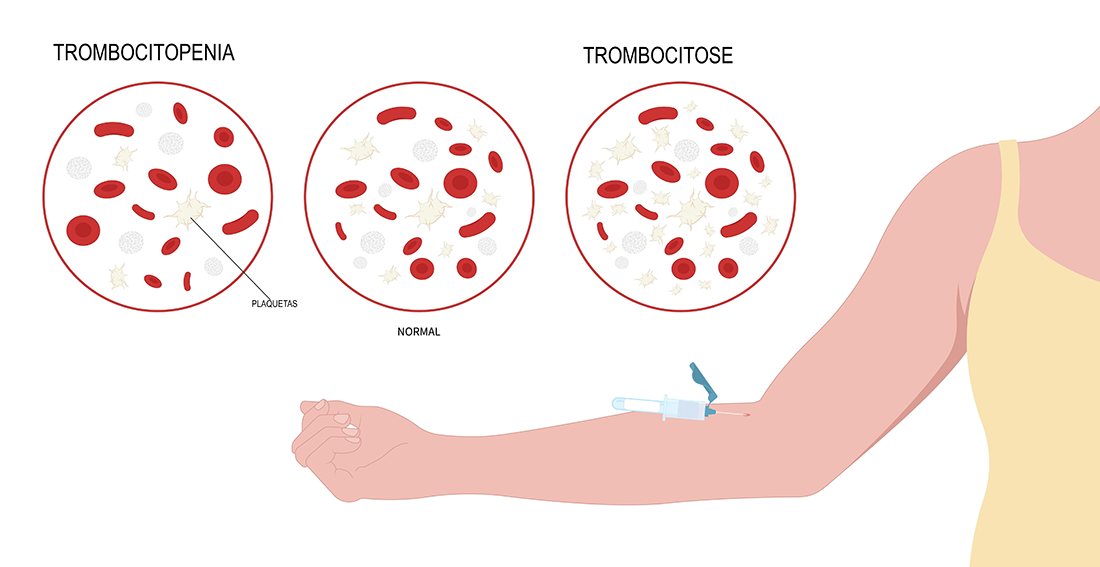
Doenças relacionadas:
- Trombocitopenia:
- Caracterizada por uma baixa contagem de plaquetas, aumentando o risco de sangramentos espontâneos. Pode incluir condições como púrpura trombocitopênica idiopática (PTI), leucemia e síndrome mielodisplásica.
- Trombocitose:
- Refere-se à contagem elevada de plaquetas, aumentando o risco de coágulos sanguíneos. Pode estar associada a distúrbios mieloproliferativos, infecções crônicas e inflamações.
- Doença de Von Willebrand (DvW):
- Distúrbio genético que afeta a função do fator de coagulação Von Willebrand, resultando em sangramentos prolongados e anormalidades na adesão plaquetária.
- Hemofilia:
- Distúrbio genético que afeta a coagulação sanguínea devido à deficiência ou ausência de fatores de coagulação, resultando em sangramentos prolongados.
- Síndrome Antifosfolípide (SAF):
- Distúrbio autoimune que pode aumentar o risco de formação de coágulos sanguíneos, afetando a função plaquetária.
- Trombose Venosa Profunda (TVP) e Embolia Pulmonar (EP):
- Condições em que ocorre a formação de coágulos sanguíneos, podendo envolver plaquetas, e que podem levar a complicações graves.
- Trombocitopatias Hereditárias:
- Distúrbios genéticos que afetam a função plaquetária, incluindo:
- Trombastenia de Glanzmann: Caracterizada por disfunção plaquetária, levando a sangramentos.
- Síndrome de Bernard-Soulier: Caracterizada por plaquetas grandes e disfuncionais.
- Distúrbios genéticos que afetam a função plaquetária, incluindo:
- Síndrome Mielodisplásica (SMD):
- Grupo de doenças da medula óssea que pode resultar em plaquetas anômalas, aumentando o risco de sangramento e infecções.
- Síndrome de Alport:
- Uma doença genética rara que pode afetar a função plaquetária, levando a distúrbios hemorrágicos.
- Trombocitopatia Amegacariocítica Congênita:
- Distúrbio genético raro que resulta em uma diminuição significativa de megacariócitos e plaquetas na medula óssea.
Exames:
Os exames relacionados às plaquetas e ao sistema de coagulação sanguínea são cruciais para diagnosticar distúrbios hemostáticos e monitorar a saúde do paciente. Aqui estão alguns exames comumente solicitados:
- Contagem de Plaquetas:
- Mede o número de plaquetas no sangue por microlitro. Ajuda a avaliar a capacidade do organismo de coagular e prevenir sangramentos ou formação excessiva de coágulos.
- Tempo de Sangramento:
- Avalia a eficácia da formação do tampão plaquetário. Um tempo prolongado pode indicar distúrbios plaquetários ou deficiências nos fatores de coagulação.
- Tempo de Tromboplastina Parcial Ativada (TTPa):
- Avalia a eficiência da via intrínseca da coagulação, ajudando a identificar distúrbios hereditários ou adquiridos nos fatores de coagulação.
- Tempo de Protrombina (TP) e Índice Internacional Normalizado (INR):
- Avaliam a via extrínseca da coagulação e são frequentemente utilizados para monitorar pacientes em tratamento com anticoagulantes.
- Dosagem de Fibrinogênio:
- Mede os níveis de fibrinogênio, uma proteína essencial para a formação de coágulos. Pode indicar distúrbios de coagulação.
- Dosagem de Fatores de Coagulação:
- Avalia os níveis de fatores específicos, como Fator VIII, Fator IX, entre outros, para diagnosticar distúrbios hemofílicos.
- Dosagem de Von Willebrand Antígeno e Atividade:
- Ajuda no diagnóstico da Doença de Von Willebrand, avaliando a quantidade e a função do fator de coagulação Von Willebrand.
- Agregação Plaquetária:
- Avalia a resposta das plaquetas a estímulos, identificando distúrbios plaquetários, como a trombastenia de Glanzmann.
- Esfregaço de Sangue Periférico:
- Permite uma análise detalhada da morfologia das células sanguíneas, incluindo as plaquetas.
- Testes Genéticos:
- Em casos de distúrbios hereditários, os testes genéticos podem ser realizados para identificar mutações genéticas associadas a condições como hemofilia ou doenças plaquetárias hereditárias.
